Predicting the Most Stable Boron Nitride Structure with Quantum Simulations
Researchers settle the debate on the relative stabilities of boron nitride's structures using a state-of-the-art quantum simulation method
Despite the huge technological interest in boron nitride (BN), understanding the relative stability of its different structural phases remains a challenge owing to conflicting results from experiments and simulations. In a new study, an international research team addresses this issue with state-of-the-art quantum Monte Carlo simulation technique, demonstrating the most stable BN structure. This accurate and reliable method could lay the groundwork for future breakthroughs in the field of materials science.
Boron nitride (BN) is a versatile material with applications in a variety of engineering and scientific fields. This is largely due to an interesting property of BN called "polymorphism," characterized by the ability to crystallize into more than one type of structure. This generally occurs as a response to changes in temperature, pressure, or both. Furthermore, the different structures, called "polymorphs," differ remarkably in their physical properties despite having the same chemical formula. As a result, polymorphs play an important role in material design, and a knowledge of how to selectively favor the formation of the desired polymorph is crucial in this regard.
However, BN polymorphs pose a particular problem. Despite conducting several experiments to assess the relative stabilities of BN polymorphs, a consensus has not emerged on this topic. While computational methods are often the go-to approach for these problems, BN polymorphs have posed serious challenges to standard computation techniques due to the weak "van der Waals (vdW) interactions" between their layers, which is not accounted for in these computations. Moreover, the four stable BN polymorphs, namely rhombohedral (rBN), hexagonal (hBN), wurtzite (wBN), and zinc-blende (cBN), manifest within a narrow energy range, making the capture of small energy differences together with vdW interactions even more challenging.
Fortunately, an international research team led by Assistant Professor Kousuke Nakano from Japan Advanced Institute of Science and Technology (JAIST) has now provided evidence to settle the debate. In their study, they addressed the issue with a state-of-the-art first principles calculations framework, namely fixed-node diffusion Monte Carlo (FNDMC) simulations. FNDMC represents a step in the popular quantum Monte Carlo simulations method, in which a parametrized many-body quantum "wavefunction" is first optimized to attain the ground state and then supplied to the FNDMC.
Additionally, the team also computed the Gibbs energy (the useful work obtainable from a system at constant pressure and temperature) of BN polymorphs for different temperatures and pressures using density functional theory (DFT) and phonon calculations. This paper was made available online on March 24, 2022 published in The Journal of Physical Chemistry C on March 24, 2022.
According to the FNDMC results, hBN was the most stable structure, followed by rBN, cBN, and wBN. These results were consistent at both 0 K and 300 K (room temperature). However, the DFT estimations yielded conflicting results for two different approximations. Dr. Nakano explains these contradictory findings: "Our results reveal that the estimation of relative stabilities is greatly influenced by the exchange correlational functional, or the approximation used in the DFT calculation. As a result, a quantitative conclusion cannot be reached using DFT findings, and a more accurate approach, such as FNDMC, is required."
Notably, the FNDMC results were in agreement with that generated by other refined computation methods, such as "coupled cluster," suggesting that FNDMC is an effective tool for dealing with polymorphs, especially those governed by vdW forces. The team also showed that it can provide other important information, such as reliable reference energies, when experimental data is unavailable.
Dr. Nakano is excited about the future prospects of the method in the area of materials science. "Our study demonstrates the ability of FNDMC to detect tiny energy changes involving vdW forces, which will stimulate the use of this method for other van der Waals materials," he says. "Moreover, molecular simulations based on this accurate and reliable method could empower material designs, enabling the development of medicines and catalysts."
Solving the relative stability conundrum is undoubtedly a huge step forward. But there is still work to be done and the pace is sure to go up!
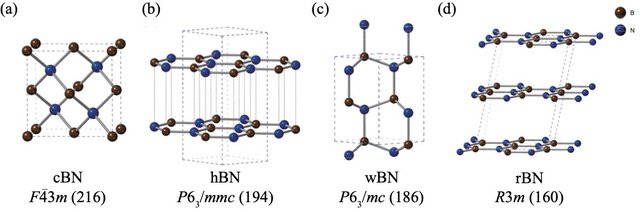
Figure 1. Crystal polymorphs of boron nitride.
The structures and space groups of (a) zinc-blende boron nitride (cBN), (b) hexagonal boron nitride (hBN), (c) wurtzite boron nitride (wBN), and (d) rhombohedral boron nitride (rBN). Boron and nitrogen atoms are depicted in brown and blue, respectively.
Image credit: Kousuke Nakano from JAIST.
Reference
| Title of original paper: | Diffusion Monte Carlo Study on Relative Stabilities of Boron Nitride Polymorphs |
| Journal: | The Journal of Physical Chemistry C |
| DOI: | 10.1021/acs.jpcc.1c10943 |
Funding information
The study was funded by a Grant-in-Aid for Early-Career Scientists (Grant Number JP21K17752), a Grant-in-Aid for Scientific Research(C) (Grant Number JP21K03400), the HPCI System Research Project (Project ID: hp210019, hp210131, and jh210045), MEXT-KAKENHI (JP16H06439, JP17K17762, JP19K05029, JP19H05169, JP21K03400, JP16KK0097, and JP19H04692), the Air Force Office of Scientific Research (Award Number: AFOSR-AOARD/FA2386-17-1-4049, FA2386-19-1-4015, and FA2386-20-1-4036), FLAGSHIP2020 (Project Nos. hp190169 and hp190167 at K-computer), and the JSPS Bilateral Joint Projects (with India DST).
April 14, 2022
